
Moleküler modelleme alanında, yanaştırma, bir molekülün ikinci bir moleküle kararlı bir kompleks oluşturmak için birbirine bağlandığında bağlandığında tercih ettiği oryantasyonu tahmin eden bir yöntemdir. Tercih edilen oryantasyon bilgisi iki molekül arasındaki birleşme veya bağlanma afinitesini tahmin etmek için kullanılabilir.
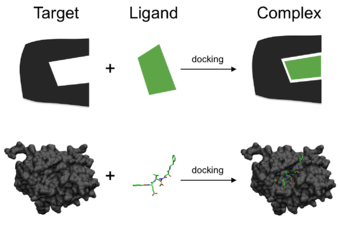
Proteinler, peptitler, nükleik asitler, karbonhidratlar ve lipitler gibi biyolojik açıdan alakalı moleküller arasındaki ilişkiler sinyal iletiminde merkezi bir rol oynar. Ayrıca, etkileşen iki partnerin nispi oryantasyonu üretilen sinyalin türünü etkileyebilir (örn. Agonizm ve antagonizm). Bu nedenle yanaştırma, üretilen sinyalin hem kuvvetini hem de tipini tahmin etmek için yararlıdır.
Moleküler yanaştırma, küçük moleküllü ligandların uygun hedef bağlanma bölgesine bağlanma-konformasyonunu tahmin etme kabiliyeti nedeniyle yapıya dayalı ilaç tasarımında en sık kullanılan yöntemlerden biridir. Bağlanma davranışının karakterizasyonu, ilaçların rasyonel tasarımında ve temel biyokimyasal süreçlerin aydınlatılmasında önemli bir rol oynamaktadır.
Dış bağlantılar
-
Bikadi Z, Kovacs S, Demko L, Hazai E. "Molecular Docking Server - Ligand Protein Docking & Molecular Modeling". Virtua Drug Ltd. 20 Kasım 2008 tarihinde kaynağından arşivlendi. Erişim tarihi: 15 Temmuz 2008.
Internet service that calculates the site, geometry and energy of small molecules interacting with proteins
- Malinauskas T. "Step by step installation of MGLTools 1.5.2 (AutoDockTools, Python Molecular Viewer and Visual Programming Environment) on Ubuntu Linux 8.04". 4 Haziran 2011 tarihinde kaynağından arşivlendi. Erişim tarihi: 15 Temmuz 2008.
- Docking@GRID31 Aralık 2019 tarihinde Wayback Machine sitesinde arşivlendi. Project of Conformational Sampling and Docking on Grids : one aim is to deploy some intrinsic distributed docking algorithms on computational Grids, download Docking@GRID open-source Linux version
- Click2Drug.org - Directory of computational drug design tools.
- Ligand:Receptor Docking with MOE (Molecular Operating Environment)