
Glial tümör, astrositomlar, ependimomlar, oligodendrogliom ve embriyonik tümörler dahil olmak üzere merkezi sinir sisteminin (MSS) sayısız tümörleri için kullanılan genel bir terimdir. Dünya Sağlık Örgütü (WHO) tümörleri ciddiyet ve nüks açısından farklı kategorilere ayırmaktadır. Bu kategorilendirme en son 2016 yılında yenilenmiştir. Derece I olarak sınıflandırılan ilk tümöre pilositik astrositom denir ve en sık çocukluk çağında görülür. Bir sonraki tümör, difüz astrositomlar derece II-III ve IV olabilirler. Düşük evreli glial tümörler zamanla evre atlayabilirler. Bu tümörler davranışsal olarak agresif yani malign özellik gösterirler.. Evre 3 astrositomlar anaplastik astrositom, evre 4 astrositom ise Glioblastom olarak isimlendirilir.
Merkezi sinir sisteminin bir diğer glial tümör tipi, ependimal tümörlerdir. Bunlar beyindeki ventriküllerin ve omuriliğin ependimal hücrelerine benzeyen hücrelerdir. Bunlar da saldırganlık derecelerine göre farklı kategorilerde sınıflandırılır. En az agresif (Evre 1) ependimal tümörler Subependimom'lar ve Miksopapiller ependimom bulunur. En şiddetli derece III olarak sınıflandırılır ve anaplastik ependimomlar olarak adlandırılır ve bunlar genellikle omurganın tabanında görülür.
Oligodendrogliom nadir görülen başka bir glial ttümördür. Normalde beynin ak maddesinde görülürler. Sebepler bilinmemekle birlikte, 1p / 19q silinmesinin pozitif olduğu vakalarda kemoterapi tedaviye yanıt daha iyidir. Oligodendroglioma, keskin sınırları ve ayırt edici "kızarmış yumurta" özellikleri nedeniyle beyin dokusundan histolojik olarak çok farklıdır.
Tarihçe
Beyin tümörleri tarihte ilk kez 1829'da bir anatomist ve patolog olan Jean Cruveilhier tarafından makroskopik olarak tanımlanmış ve 1836'da Bressler tarafından sınıflandırılmıştır. Tarihte Gliom terimini ilk kez kullanan kişi Rudolf Virchow'dur. Harvey Williams Cushing ve Jean Cruveilhier 1926 yılında Tumors Arising from the Blood-Vessels of the Brain isimli yayınlarında ilk sistematik sınıflandırmayı yaptılar. Ancak bu sınıflandırma karmaşık olduğu için ilgi görmedi. Yirminci yüzyılın ortalarında James Watson Kernohan yeni basit bir sınıflandırma geliştirdi. Karmaşık histogenetik sınıflandırmayı: Astoristom, ependimom, nöroastrositom, medulloblastom, oligodendrogliom olarak 5 glial tümör kategorisine indirdi. Ayrıca bu tümörlerin 4 derece şeklinde gruplandırma sistemini geliştirdi. Bu sistem günümüzde kullanılan sistemin temellerini atmış oldu. İlk kez Dünya Sağlık Örgütü (DSÖ) 1979'da uluslararası olarak tümörlerin sınıflandırmasını yapmıştır. Bu sınıflandırma yine Dünya Sağlık Örgütü (DSÖ) tarafından 1993'te revize edilmiştir. 2000 yılında Dünya Sağlık Örgütü (DSÖ) standardizasyonu sağlamak, bazı moleküler ve genetik bilgileri kapsamak, ICD hastalık tanımlama kodlarını içerecek, tanısal kriterleri belirlemek amacıyla sınıflamayı revize etti. 2007 yeni yılında yine DSÖ tarafından tüm tümörler 7 ana gruba ve 13 alt gruba ayrıldı. 2014 yılında ISN (International Society of Neuropathology) tarafından yapılan toplantıda (HAARLEM 2014) DSÖ'nün en 2007 yılında yaptığı sınıflamanın tekrar gözden geçirilmesi gerektiği, genetik ve moleküler bilgilerin gözden geçirilmesi önerildi. Ayrıca genetik/moleküler bilgilerin tanıda kullanımını öngören entegre tanı kavramını ortaya attılar. Mevcut sınıflandırma aşağıda açıklandığı gibi en son DSÖ tarafından 2016 yılında güncellendi.
Dünya Sağlık Örgütü (WHO) 2016 Merkezi Sinir Sistemi tümörlerinin sınıflandırılması
Dünya sağlık örgütü santral sinir sistemi tümörlerinin sınıflandırmasını en son 2016 yılında yapılmıştır.. En son 2007 yılında yapılmış olan bu sınıflamaya 2016 yılında Histolojik değerlendirmelere ek olarak artık moleküler parametreler de eklendi. Bu yeni sınıflamayla birlikte bazı tümör tanımlamaları kaldırıldı, yeni tümör tanımlamaları eklendi. Geçen yüzyılda beyin tümörlerinin sınıflandırılması tümör hücrelerinin mikroskop altındaki görünümleri ve hücresel düzeydeki farklılıkları ile yapılırdı.
Son 20 yılda tümör oluşumundaki genetik faktörlerin, aynı tümör içinde bile genetik farklılıkların olduğunu ve bunların tedavide ve sağ kalımda farklılıklar gösterdiği ortaya konuldu. Bu sayede yeni tümör alt tiplerinin varlığı ortaya konuldu. Bu nedenele 2016 yılında yapılan yeni sınıflandırma ile yüzyıldır uygulanan ve tümör tiplendirilmesinde kullanılan ışık mikroskopik incelemere genetik/moleküler incelemeler eklenmiş oldu. 20 ülkeden 117 katılımcının bulunduğu, 3 gün süren konsensusun ardından 2016 sınıflandırılması yayınlandı.
Sınıflandırma
| Diffüz astrositom, ''IDH-mutant'' | |||
| Gemistositik astrositom, ''IDH-mutant'' | |||
| Diffüz astrositom, ''IDH- wild tip'' | |||
| Diffüz astrositom, ''NOS'' | |||
| Anaplastik astrositom | IDH-mutant | ||
| IDH-wilt tip | |||
| NOS | |||
| Glioblastom, ''IDH wild-tip'' | Dev hücreli glioblastom | ||
| Gliosarkom | |||
| Epiteloid glioblastom | |||
| Glioblastom | IDH mutant | ||
| NOS | |||
| Diffüz Orta Hat Gliomu (midline glioma) | H3 K27M-mutant | ||
| Oligodendrogliom | IDH-mutant ve 1p-19q co-deletion | ||
| Oligodendrogliom | NOS | ||
| Anaplastik oligodendrogliom | IDH-mutant ve 1p/19q co-deletion | ||
| Anaplastik oligodendrogliom | NOS | ||
| Oligoastrositom | NOS | ||
| Anaplastik oligoastrositom | NOS | ||
| Diğer astrositomlar | Diğer astrositik tümörler | Pilositik astrositom | |
| ''Pilomiksoid astrositom'' | |||
| Subependimal dev hücreli astrositom | |||
| Pleomorfik ksantoastrositom | |||
| Anaplastik pleomorfik ksantoastrositom | |||
| Ependimomlar | Ependimal tümörler | Subependimom | |
| Miksopapiller ependimom | |||
| Ependimom | Papiller pendimom | ||
| Berrak hücreli ependimom | |||
| Tanisitik ependimom | |||
| Ependimom, RELA-füzyon pozitif | |||
| Anaplastik ependimom | |||
| Diğer gliomlar | 3. ventrikülün koroid gliomu | ||
| Anjiojenik gliom | |||
| Astroblastom | |||
| Embriyonel tümörler | Medulloblastom (genetik tanımlama) | Medulloblastom, WNT-aktif | |
| Medulloblastom, SHH-aktif ve TP53 mutant | |||
| Medulloblastom, SHH aktif ve TP53-wild tip | |||
| Medulloblastom, non-WNT/non-SHH grup 3 ve grup 4 | |||
| Medulloblastom (histolojik tanımlama) | Klasik tip | ||
| desmoplastik/boduler | |||
| yaygın nodüler tip | |||
| dev hücreli/anaplastik | |||
| Medulloblastom, NOS | |||
| Çok tabakalı rozetlerden oluşan embriyonel tümör | C19MC-değişken | ||
| Çok tabakalı rozetlerden oluşan embriyonel tümör | NOS | ||
| Medulloepitelyoma | |||
| Nöroblastom | |||
| Ganglionöroblastom | |||
| Embriyonel Tümör | NOS | ||
| Atipik teratoid/rabdoid tümör | |||
| Rabdoid özellik gösteren santral sinir sistemi
embriyonel tümörü |
|||
| Kranial ve paraspinal sinir tümörleri | Schwannom | Sellüler Schwannom | |
| Pleksiform Schwannom |
| Tümör | alt tip | Evre |
|---|---|---|
| Diffüz astrositik ve oligodendroglial tümörler | Diffüz astrositom, IDH mutant | II |
| Anaplastik astrositom, IDH mutant | III | |
| Gliblastom, IDH-Wild tip | IV | |
| Glioblastom, IDH-mutant | IV | |
| Diffüz orta hat gliomu, H3 K27M-mutant | IV | |
| Oligodendrogliom, IDH-mutant, 1p-19q co-deltion | II | |
| Anaplastik oligodendrogliom, IDH mutant ve 1p-19q co-deletion | III | |
| diğer astrositik tümörler | Pilositik astrositom | I |
| Subependimal dev hücreli astrositom | I | |
| Pleomorfik ksantoastrositom | II | |
| Anaplastik pleomorfik ksantoastrositom | III | |
| ependimal tümörler | Subependimom | I |
| Miksopapiller ependimom | I | |
| Ependimom | II | |
| Ependimom RELA-fusion pozitif | II-III | |
| Anaplastik ependimom | III | |
| Diğer gliomlar | Anjiosentrik gliom | I |
| 3. ventrikülün kordoid tümörü | II | |
| Nöronal veya nöronal-glial tümörler | Disembriyoplastik nöroepitelyal tümörler | I |
| Gangliositom | I | |
| Gangliogliom | I | |
| Anaplastik gangliogliom | III | |
| Serebellumun displastik gangliositomu (Lhermitte_Duclos) | I | |
| Desmoplastik infantil astrositom ve gangliogliom | I | |
| Papiller glionöronal tümör | I | |
| Rozet oluşturan glionöronal tümör | I | |
| Santral nörositom | II | |
| Ekstraventriküler nörositom | II | |
| Serebellar liponörositom | II | |
| Embriyonel tümörler | Medulloblastom (tüm alt tipler) | IV |
| Embriyonel tümör, çok katlı rozet oluşturan, C19MC-altered | IV | |
| Medulloepitelyoma | IV | |
| Santral sinir sistemi embriyonel tümörü, NOS | IV | |
| Atipik teratoid/rabdoid tümör | IV | |
| Rabdoik özellik gösteren santral sinir sistemi embriyonel tümörü | IV | |
| Kranial ve paraspinal sinirlerin tümörleri | Schwannom | I |
| Neurofibrom | I | |
| Perinöroma | I | |
| Malin periferik sinir kılıfı tümörü (MPNST) | II, III, IV | |
| Evre II: Hücresel atipi, Evre III: Artmış mitoz, Evre IV: mikrovasküler proliferasyon ve nekroz | ||
Diffüz astrositik ve oligodendroglial tümörler
Difüz astrositomlar ve Anaplastik astrositomlar
Difüz gliomlar Dünya sağlık örgütünün en son 2016 yılında tanımladığı, birçok farklı alt tipe sahip glial tümörün genel ismidir. Beynin kendi dokusu olan glial hücrelerden kaynaklanan tümör alt tiplerinden biridir. Daha önceki glial tümör sınıflandırılması sadece histopatolojik incelemeye göre yapılırken son sınıflamada moleküler ve genetik incelemeler sınıflamaya dahil edilmiştir. Artık hem astrositik hem de oligodendroglial tümörler bu sınıflamaya dahil edilmiştir. Bunun nedeni büyüme paternlerinin benzerlik göstermesiyle birlikte benzer genetik mutasyonlar göstermelerinden kaynaklanmaktadır. Bu sınıftaki tümörler hem fenotipik hem de genetik özelliklerine göre sınıflandırılmıştır. Hastalığın seyrinin belirlenmesi, genel veya hedefe yönelik tedavilerin belirlenmesi açısından bu sınıflamanın önemi çok fazladır.
Son sınıflamaya göre Difüz gliomlar, derece II ve III astrositik tümörler, derece II ve III oligodendroglial tümörler, derece IV glioblastomlar ve çocukluk çağı difüz gliomlardan oluşur. Işık mikroskobu altında yapılan incelemelerde; derece II artmış hücresel atipi, derece III atipi ile birlikte artmış mitoz, derece IV ise öncekilere ek olarak mikrovasküler proliferasyon ve nekroz özelliği gösteren tümörlerdir.
WHO derece II astrositomlar ve WHO derece III anaplastik astrositomlar ayrı ayrı IDH-mutant, IDH-wild tip (vahşi tip) ve NOS kategorilerine ayrıldı. Eğer IDH testi yapılabiliyorsa derece II ve derece III tümörler çoğunlukla IDH-mutant'tır. Eğer immünohistokimyasal olarak R132H IDH1 codon 132 ve IDH2 codon 172 gen proteinleri negatif ise veya IDH1 codon 132 ve IDH2 codon 172 genlerinden herhangi biri negatif ise mevcut lezyon IDH-wild (vahşi) tip olarak sınıflandırılır. Akılda tutulmalıdır ki IDH-wild tip diffüz astrositom nadir konulan tanılardan biridir. IDH mutasyonunun varlığı yüksek dereceli gliomlarda (Derece III-IV) prognozun belirlenmesinde histolojik dereceden ve diğer moleküler özelliklerden daha önemlidir. IDH mutasyonu varlığı durumunda Derece III difüz astrositom derece II gibi davranabilirken, IDH mutasyon yokluğunda derece IV olan glioblastom gibi davranabilir. Ancak gliom klasik tedavi protokollerinde IDH mutasyon durumunun tedaviye yanıtta etkisi olmadığı gösterilmiştir. 2016 yılında yapılan son güncellemede önceki sınıflamada olan protoplazmik astrositom ve fibriler astrositom sınıflandırmadan çıkarılmıştır.
Gliomatosis cerebri son sınıflandırmadan çıkarılmıştır. Bunun nedeni aslında bu durumun ister IDH mutant ister IDH-wild tip olsun tüm alt sınıflarda gözlenebilen bir büyüme paterni olmasından kaynaklanmaktadır.gliomatosis serebri terimi bir tümör sınıflandırmasından ziyade artık tüm tümör tipleri için tümörün üç ve daha fazla loba yayılması anlamında kullanılmaktadır.
Glioblastom
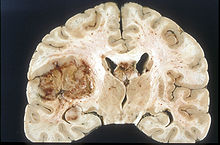

Glioblastomlar derece IV tümörler olarak tanımlanır. Daha önceki sınıflandırmada sadece histopatolojik değerlendirme ile sınıflandırılırken, son sınıflamada IDH mutasyonunun varlığı gibi moleküler farklar gösterilmiştir. Bu farklılıklar Glioblastomun kendi alt tiplerinde prognozun belirlenmesinde oldukça büyük öneme sahiptir. Glioblastomlar WHO 2016 sınıflandırmasında iki ana başlık altında değerlendirilmektedir. Birincisi en sık 55 yaş üstünde görülen, de-novo glioblastom olarak da bilinen, vakaların %90'ını oluşturan IDH-wild (vahşi) tip glioblastomdur. Önceki tanımlamada bu tümör primer glioblastom olarak isimlendirildi. IDH-wild tip glioblastomların alt sınıfı olan dev hücreli glioblastom ortalama olarak daha uzun sağ kalıma sahiptir (13 ay). Yine bu tipin bir alt varyantı olan epiteloid glioblastom ise yeni tanımlanmıştır. Daha çok gençlerde görülen bu tip daha kötü prognoza sahiptir. IDH-wild tip tüm glioblastomların yaklaşık %90 kadarını oluşturur. Yıllık insidansı 3-4/100000'dir. Median yaş 62 olup erkeklerde daha sık görülür.
Daha önce sekonder glioblastom olarak bilinen ve glioblastomların diğer alt tipi olan IDH-mutant glioblastom ise vakaların kalan %10'unu oluşturan, daha önceden düşük dereceli difüz gliom olarak bilinen ve daha genç yaşlarda ortaya tipidir. Eğer tam IDH testi yapılamıyorsa glioblastom, NOS olarak sınıflandırılır. Median yaş 45 olup kadın-erkek insidansı eşittir. Daha sıklıkla frontal bölgeyi tutar.
İlkel nöronal komponenti olan Glioblastom (Glioblastoma with primitive neuronal component sınıflandırmaya dâhil edilmiştir. Bu tanımlama daha önce PNET benzeri içeriğe sahip glioblastom (Glioblastoma with PNET-like component) olarak bilinmekteydi. Bu tanımlama diffüz astrositomlar hatta nadir olarak oligodendrogliomlar ile karışabilmekteydi. %25'i düşük dereceli gliomlardan gelişir. Bu sınıflamanın önemi, bu alt tipteki tümörlerde Beyin omurilik sıvısı yoluyla omuriliğe yayılım görülebileceğinden dolayı takiplerde omuriliğin de takip edilmesi önerilir.
Oligodendrogliom
Oligodenrogliomlar WHO 2016 sınıflamasına göre derece II veya derece III tümörler olarak sınıflandırılırlar. Derece II olan oligodendrogliom ve derece III olan anaplastik oligodendrogliom ayrımı, IDH mutasyonu ile birlikte 1p/19q genlerinin ikisinin birden delesyon varlıklarına göre yapılır. IDH-mutant sınıflaması için IDH1 codon 132 ve IDH2 codon 172 genlerinin her ikisinde de mutasyon olmalıdır. Eğer genetik test yapılamıyorsa veya anlamlı sonuç alınamıyorsa Oligodendrogliom, NOS olarak isimlendirilmelidir. IDH mutant oligodendrogliomlar tedaviden bağımsız olarak uzun sağ kalıma sahiptirler.
Oligoastrositom
Oligoastrositom 2016 WHO sınıflaması oldukça problemlidir. Bunun nedeni histolojik özellik olarak neredeyse tüm astrositik ve oligodendroglial varlık gösteren tümörler genetik testler kullanıldığında astrositom veya oligodendrogliom olarak değerlendirilebilirler. Derece II oligoastrositom ve derecce III anaplastik oligoastrositom tanısı NOS tanımalamasına bağlıdır ve ancak uygun tanısal moleküler testlerin negatif olduğu durumlarda tanı alabilir. Literatürde oligodendrogliom ve astrositom komponentinin her ikisinin fenotipik ve genetik özelliklerinin birlikte bulunduğu gerçek oligoastrositomlar nadir olarak bulunur.
Diffüz orta hat gliomları
Geçmişte çocukluk çağı gliomlar, davranışsal farklarına rağmen erişkin tip gliomlar gibi sınıflandırılırdı. Altta yatan genetik farklılıklardan dolayı çocukluk çağı difüz gliomları erişkin tiplerinden ayrılırlar. Sınıflandırılmış gruplardan biri çocukluk çağında (nadir olarak erişkinde de izlenebilir) bir Histon geni olan H3 H3F3A'da K27M mutasyonu izlenir. Bu yapıya sahip tümörler yaygın büyüme paterni, orta hat yerleşimi (talamus, beyin sapı, omurilik) gibi özellikler gösterirler. Daha önce Difüz intrinsik pontin gliom (DIPG) olarak tanımlanan bu tümör artık Difüz Orta Hat gliomu, H3 K27M-mutant olarak isimlendirilmektedir.
Diğer astrositomlar
2016 WHO sınıflamasında daha önce neoplastik özellik gösteren Pleomorfik ksantoastrositom olarak bilinen tümör artık Anaplastik pleomorfik ksantoastrositom, WHO grade III olarak sınıflandırılmaktadır. Pleomorfik ksantorastrositomu anaplastik olarak sınıflandırmak için ışık mikroskobunda 10 kat büyütmede sahada beş ve daha fazla mitoz izlenmesi gereklidir. Nekroz izlenebilir ama mitozun düşük olduğu durumlarda nekrız varlığının anlamı tartışmalıdır. Anaplatik alt tipte sağ kalım derece II tümöre göre kısadır.
pilomiksoid astrositom derecelendirilmesi de 2016 WHO sınıflandırmasında değişmiştir. daha önce WHO derece II olarak sınıflandırılmış olan bu tümör, bir varyantı olan pilositik astrositomlardan farklı olarak, özellikle erişkinlerde daha agresif seyir göstermektedir. Bu nedenle pilomiksoid astrositom sınıflandırılmasının direkt olarak derece II olması konusu halen tartışmalıdır.
İyi sınırlı tümörlerdir. IDH gen değişikliği göstermezler. Sıklıkla BRAF (PA ve PXA) ve TSC1/TSC2 (SEGA) gen mutasyonu gösteren gliomlar difüz gliomlardan ayrılarak diğer astrositik tümörler grubu içine girerler.
Ependimomlar
Daha önceki ependimomlarda ki sınıflandırma klinik kullanımda zorluklara sebep olduğu gibi prognozun belirlenmesinde ve tedavi algoritmalarının oluşturulmasında faydası olmadığı biliniyordu. Beklenti daha ileri çalışmalarla genetik olarak ependimomların daha fazla sınıflandırılması yönünde. Son sınıflandırmada kabul edilen tek genetik alt tip Ependimom, RELA fusion-pozitiftir. Bu tümör tipi çocukluk çağı supratentorial tümörlerin büyük çoğunluğunu oluşturmaktadır. L1CAM proteinin ekspresyonundaki artış bu tümör için uygun bir immuno-histokimyasal tetkik olabilir. Daha önce mevcut olan ve standart ependimomlar ile karışan selüler ependimomlar sınıflandırmadan çıkarılmıştır.
Ependimom RELA-füzyon pozitif sınıflandırmaya yeni eklenen ve moleküler tiplendirmenin esas alındığı ilk ependimomdur. DSÖ derece II-III tümörlerdir. Daha çok çocuklarda ve supratentoryal yerleşimli tümörlerdir. Genetik olarak C1 10rf95-RELA olarak tanımlanır. RELA bir NFĸβ sinyal yolunda bir transkripsiyon faktörüdür. Kötü prognoza sahiptir.
Nöronal ve nöronal-glial tümörler
Yeni tanımlanmış Difüz leptomeningeal glionöronal tümör daha önce literatürde farklı benzer isimli tümörlerle anılmış olsa da en sık Çocukluk çağının oligodendroglial benzeri leptomeningeal tümörü olarak var olmuştur. Bu tümör daha çok omurilikte olmak üzere parankimal komponenti olmayan difüz leptomeningeal tutulum gösterir. Daha çok çocuk ve adolesanlarda görülür. Bu tümör grubu IDH mutasyonu göstermez. Prognoz değişkenlik gösterir. Yavaş büyüyen bir tümör olmasına rağmen hidrosefaliye bağlı komplikasyonlar izlenebilir.
Histolojik olarak nöronal diferansiyasyon ve hem nöronal hem de glial diferansiyasyon gösterebilirler. Nöronal diferansiasyonu gösteren morfolojik özellikler neoplastik nöronlar, ganglioid hücre morfolojisi ve nörositik rozetlerdir. Glial diferansiasyon ise astrositik, oligodendroglial, ependimal difreransiasyonun morfolojik olarak belirlenmesiyle tanınabilir.
Ganglion hücreli tümör ile benzerlik gösteren, multinodüler ve vakuollu yapıya sahip yeni bir alt tip tanımlanmıştır. Serebrumun multinodüler ve vakuollü tümörü olarak sınıflandırılmıştır. Düşük dereceli tümörlerdir, glial ve nöronal farklılaşmalar gösterebilirler. İyi prognoz gösterirler.
Medulloblastom


Medulloblastomlar histolojik ve genetik olmak üzere iki şekilde sınıflandırılırlar. Histolojik olarak: desmoplastik/nodüler, nodüler yayılım, dev hücreli, anaplastik sınıflandırılır. Genetik (moleküler) olarak ise: WNT-aktif, SHH-aktif ve numerik olarak Grup 3 ve Grup 4. Bu histolojik ve genetik alt tipler dramatik olarak sağ kalım farklılıkları gösterirler.
Medulloblastom sınıflandırılması
| Genetik profil | Histoloji | Prognoz |
|---|---|---|
| Medulloblastom, WNT-akrif | Klasik
Büyük hücreler / anaplatik (nadir) |
Düşük riskli tümör, tüm WNT aktif tümörlerde aynı klasik morfolojik
yapı bulunmaktadır. |
| Medulloblastom, SHH-aktif,
TP53-mutant |
Klasik
Büyük hücreler/ anaplastik Desmoplastik/nodüler (çok nadir) |
Nadir görülen yüksek riskli tümör
En sık 7-17 yaş arasında izlenir |
| Medulloblastom, SHH-aktif,
TP53-wild tip |
Klasik
Büyük hücre/anaplastik Desmoplastik/nodüler Artmış nodülarite |
Standart riskli tümör
İnfantlarda düşük riskli tümör |
| Medulloblastom, non-WNT, non-SHH
Grup 3 |
Klasik
Büyük hücre/anaplastik |
Standart risk
Yüksek riskli tümör |
| Medulloblastom, non-WNT, non-SHH
Grup 4 |
Klasik
Büyük hücre /anaplastik (nadir) |
Standart risk
Bilinmeyen klinikopatolojik farklılık |
WNT alt tip
|
SHH alt tip
|
Grup 3 MB/non-WNT, non-SHH
|
Grup 4 non-WNT, non-SHH
|
Diğer embriyonal tümörler
Medulloblastom dışındaki embriyonel tümörlerden Primitif nöroektodermal Tümör (PNET) tanımı kaldırılmıştır.. Yeni sınıflama 19. kromozomun (19q13.42) C19MC bölgesinin amplifikasyonuna göre yapılmıştır. Daha önceden ETANTR, ependimoblastom, medulloepitelyoma şeklinde isimlendirilen tümörler artık C19MC-amplifiye tümörler olarak sınıflandırılmaktadır.
Atipik teratoid/rabdoid tümörler (AT/RT) artık INI1 ve daha nadir olarak BRG1 gen mutasyonlarına göre sınıflandırılmakta. Bu genlerdeki farklılıklar karşılığı olan proteinlerin immünohistokimyasal olarak ölçülen değişiklikleri üzerinden değerlendirilmektedir. Eğer bir tümör histolojik olarak AT/RT gibi görünüp, genetik değişiklikler tespit edilemez ise merkezi sinir sisteminin rabdoid özzellik gösteren embriyonel tümörü (CNS embriyonal tumor with rhabdoid features olarak isimlendirilir. Sonuç olarak AT/RT tanısı konabilmesi için genetik mutasyonun gösterilmesi şarttır.
Sinir kılıfı tümörleri
Sinir kılıfı tümörleri sınıflandırılması 2007 sınıflandırılması ile ufak farklar dışında benzerlik gösterir. Melanositik schwannom artık klasik schwannomlardan tamamen ayrı bir sınıf olarak değerlendirilmektedir. Hibrit sinir kılıfı tümörleride 2016 sınıflandırmasına dahil edilmiştir. malin periferik sinir kılıfı tümörlerine (MPNST) iki yeni alt tip eklenmiştir: ""Epiteloid MPNST ve perinöral farklılaşma gösteren MPNST.